I have two files with about 4 fasta sequences each in the PHYLIP format for Da/Ds (Ka/Ks) comparison. After a search on the net, I found that the best software to do so is PAML and it requires a control file and 2 input files. For the tree input, I used PHYML which accepts PHYLIP file inputs and gives NEWICK format outputs. However I am unsure as to how to get the sequence file. I tried the sequential format but it gives me the "Error in sequence data file" error.
This is my control file. Here is my sequence file and here is my tree file.
Could I get some advice on what is wrong with my input file please. Also, this is just one set of the sequences which I wanted to compare. How do I compare two sets of sequences for the Da/Ds values. I know that this might be basic so thank you for your patience.
I don't think your sequence file is in correct format and does not look like a multiple alignment. You need a multiple sequence alignment as input. You can use the online tools (either Readseq or Built-in converter) at Phylogeny.fr website to check if your sequence file is in correct format (by trying to convert to other format).
The sequences in your file are also not of same length. If the 'cleandata' option is set to 1 in the .ctl file (which is the default), PAML will remove any sites with gaps in at least one sequence.
The way that ambiguity characters and alignment gaps are treated in baseml and codeml depends on the variable cleandata in the control file. In the maximum likelihood analysis, sites at which at least one sequence involves an ambiguity character are removed from all sequences before analysis if cleandata = 1, while if cleandata = 0, both ambiguity characters and alignment gaps are treated as ambiguity characters. In the pairwise distance calculation (the lower-diagonal distance matrix in the output), cleandata = 1 means "complete deletion", with all sites involving ambiguity characters and alignment gaps removed from all sequences, while cleandata = 0 means "pairwise deletion", with only sites which have missing characters in the pair removed.
ADD COMMENT
• link
updated 2.2 years ago by
Ram
43k
•
written 9.5 years ago by
Siva
★
1.9k
0
Entering edit mode
FYI: "'cleandata' option is set to 1 in the .ctl file (which is the default)" - actually the default is cleandata=0 (see PAML doc, page 25)
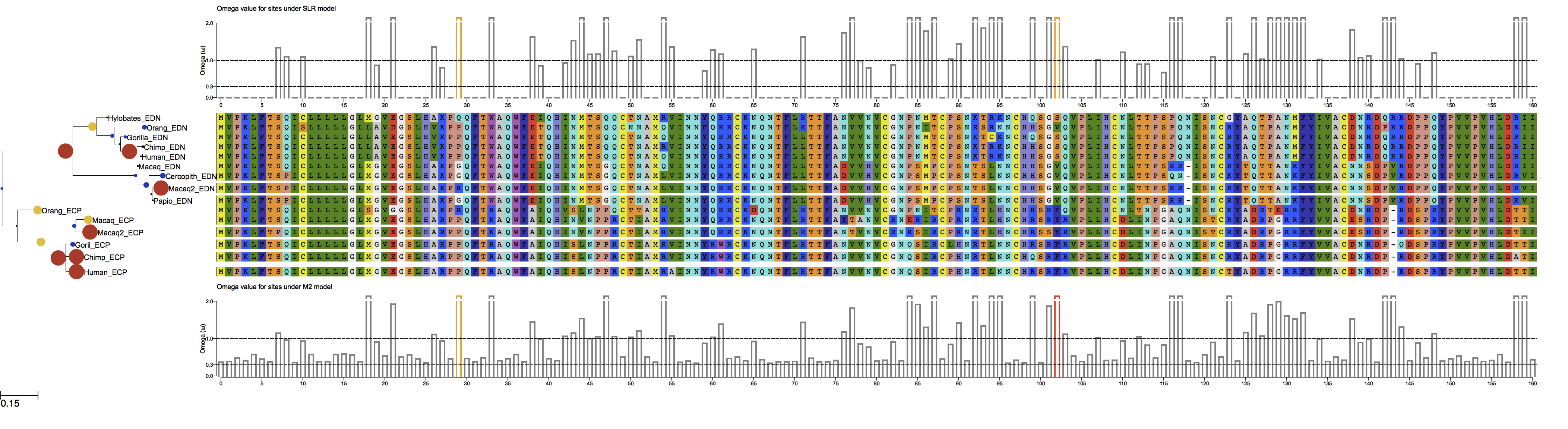
ete-evol is a wrapper tool for codeml and SLR. It simplifies its usage and produces graphical representation of results. Your tree and alignment files could be passed directly to the program, and the branches to test could be marked interactively. It also allows to run various models at once and produces figures for better interpretation of results:
ADD COMMENT
• link
updated 4.3 years ago by
Ram
43k
•
written 8.3 years ago by
jhc
★
3.0k
You want to make sure that your sequences are the same length. These sequences are clearly not aligned. Recall that an alignment is an inference of homology, so please make sure that this file represents the same protein across samples before performing an alignment.
The phylip file format's first line requires two numbers: the number of samples and the length of the samples. Since your samples have different sequence lengths, your input is in violation of the phylip standard. Your tree, in Newick (parenthetical) format, appears to be fine.
These estimates of dN/dS (omega) are maximum likelihood estimates from your alignment - your control file is setup to estimate a one-rate (global) analysis, but you can also get an estimate per position by fitting other models and selecting among them. I'm not sure what you mean by "comparing two sets of sequences." This analysis is performed per-alignment, and the power to infer patterns of selection comes from your overall sequence divergence (i.e., the structure of your tree).





FYI: "'cleandata' option is set to 1 in the .ctl file (which is the default)" - actually the default is cleandata=0 (see PAML doc, page 25)