Entering edit mode

8.9 years ago
manetsus
▴
40
AFIK, Short sequencing and short pairwise sequencing generated by next-generation-sequencing is common now-a-days and there exists several alignment tools for them.
But I want to ask about long pairwise sequencing. There exists some alignment tools for long sequences like rHAT. That means there is at least one tool which generates long reads.
Now, I am asking that is there any tool which generates Long Pairwise Reads? if yes, then is there exist any aligner or mapper for that? Give me some idea about it.
Thanks in advance.

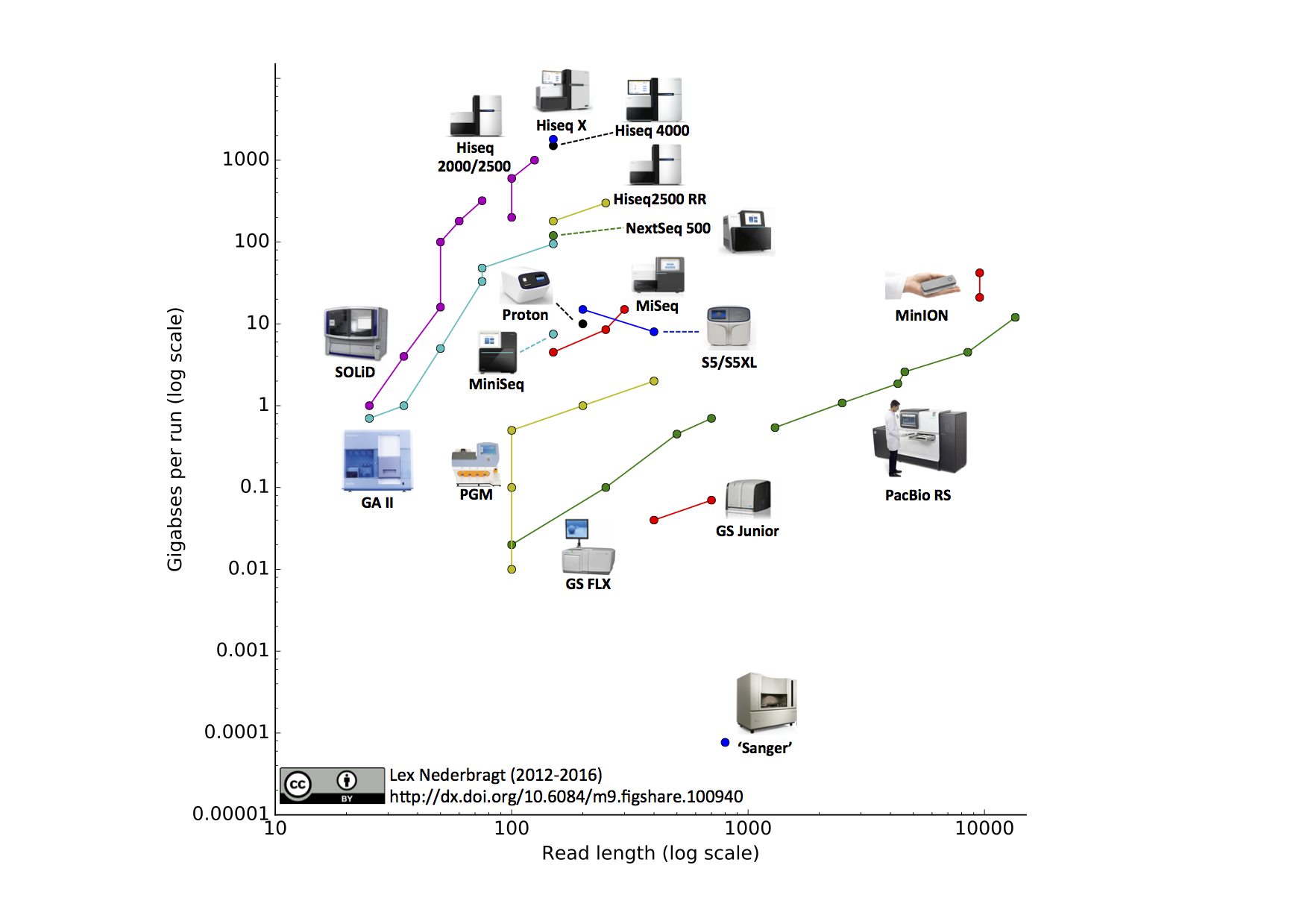

If your reference to
pairwise readsis referring to reading both strands of DNA at the same time .. then no such technology exists (as noted below Oxford nanopore/Pacbio can read both DNA strands sequentially, but not at the same time). In addition there is no need for this since DNA strands are anti-parallel complements of each other so once you sequence one strand the corresponding base on the other strand can be inferred.For oxford nanopore you can also read the other strand for consensus calling and higher quality. It was more important with older chemistry, but it's still an improvement with current chemistry.
By pairwise reads, I meant k+d+k length reads where d is the gap between two elements of the pair. Each element consist of length k. In case of computation which is written separating a
|sign. This problem may clear a little bit more about the pairwise reads. Thanks a lot for your time.This is a bit of a nitpick but you should call these "paired end" reads, a more accurate representation of the scenario you described. A single fragment being sequenced from two ends.
The word "paied" is unknown to me and I found nothing by googling. Is this a typo?
It was a typo. Thanks for the reminder.
Where d presumably is greater than the normal insert size for an Illumina NGS library?