Entering edit mode

8.0 years ago
biomagician
▴
410
Dear all,
I have indexed the C. elegans reference genome with:
bwa index output/genome/ref/seq/celegans.fa
and then aligned my de novo assembly to the reference with:
bwa mem -t 8 -x intractg output/genome/ref/seq/celegans.fa input/assembly/celegans/hgap/bristol/assembly.fa > output/alignment/pacbio/bwa/ref/bristolAssembly.sam
I then
samtools view -bS output/alignment/pacbio/bwa/ref/bristolAssembly.sam > output/alignment/pacbio/bwa/ref/bristolAssembly.bam
samtools sort output/alignment/pacbio/bwa/ref/bristolAssembly.bam -o output/alignment/pacbio/bwa/ref/bristolAssemblySorted.bam
samtools index output/alignment/pacbio/bwa/ref/bristolAssemblySorted.bam
to visualise the alignment in IGV.
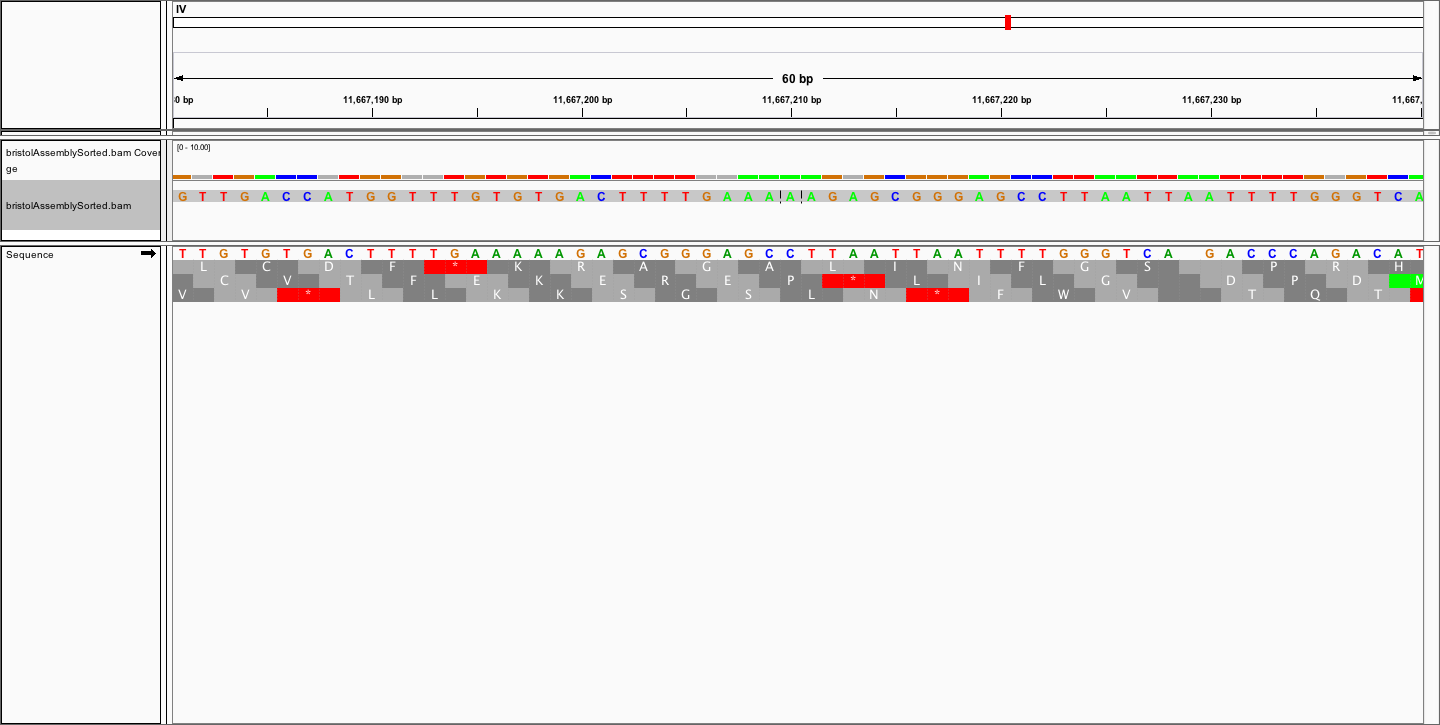
This is what I get: http://cristian-riccio.ch/wp-content/uploads/2017/07/igv_snapshot.png
{kind=link}
You can see that my assembly is the same as the reference, just shifted by 12 bases or so. Has anyone any suggestions about how to solve this problem?
Thanks.
Best,
C.
{kind=link}
Have you tried to use the actual reference sequence you used for your own alignments (instead of going on the label of genome build) in IGV?
Thanks, good idea. I've tried but there is no change. I went on the Genomes tab in IGV, loaded the reference I used for alignment and then loaded my BAM file. Still the same.
Hmm. You are actually aligning the assembly not the original reads. Assuming the assembly is long, have you tried to use a tool meant for such alignments as blat, LAST, or LASTZ? Since this is C. elegans, even mauve may be able to handle the alignments with the added benefit of showing you any rearrangements.
genomax, do you know if Mauve can output a BAM file?
is it really shifted, or just IGV is showing you this 12 bp shift?
It is really shifted I think. On this image, I am only showing the mismatched bases and you can see that there are many mismatches despite the two sequences being the same. http://cristian-riccio.ch/wp-content/uploads/2017/07/igv_snapshotreallyshifted.png
I'm asking about the BAM file, not the IGV visualization. If it's a true missalignment, then you can calculate from the BAM fields (like NM) the number of differences, and from the CIGAR string you can track down where these mismatches could be if truly they exist