I want to understand -
How sleuth is different from ballgown when we do differential gene expression analysis [apart from alignment free vs alignment based reasons].
When i compared both of them, i got reasonably good number of DGEs with sleuth, however, with ballgown i didn't get any differentially expressed gene (q-value <=0.05 for both). Please explain this. Can you we rely on Fold changes even if we don't have significant q-value?
Hi, what do you mean by "apart from alignment free vs alignment based reasons" ? Actually that makes lot of difference depending on the type of RNA/genes you are looking for. You can see this recent article where they explain it in detail.
Can you we rely on Fold changes even if we don't have significant q-value?
If you have good number of replicates in each group, I would still prefer to fix some cut-off with q-value/FDR (not the regular 0.05 but little lenient for FDR/q-value and strict in LogFC cutoff).
If you still like to use only logFC as cut-off, have a look at GFOLD.
Can you we rely on Fold changes even if we don't have significant q-value?
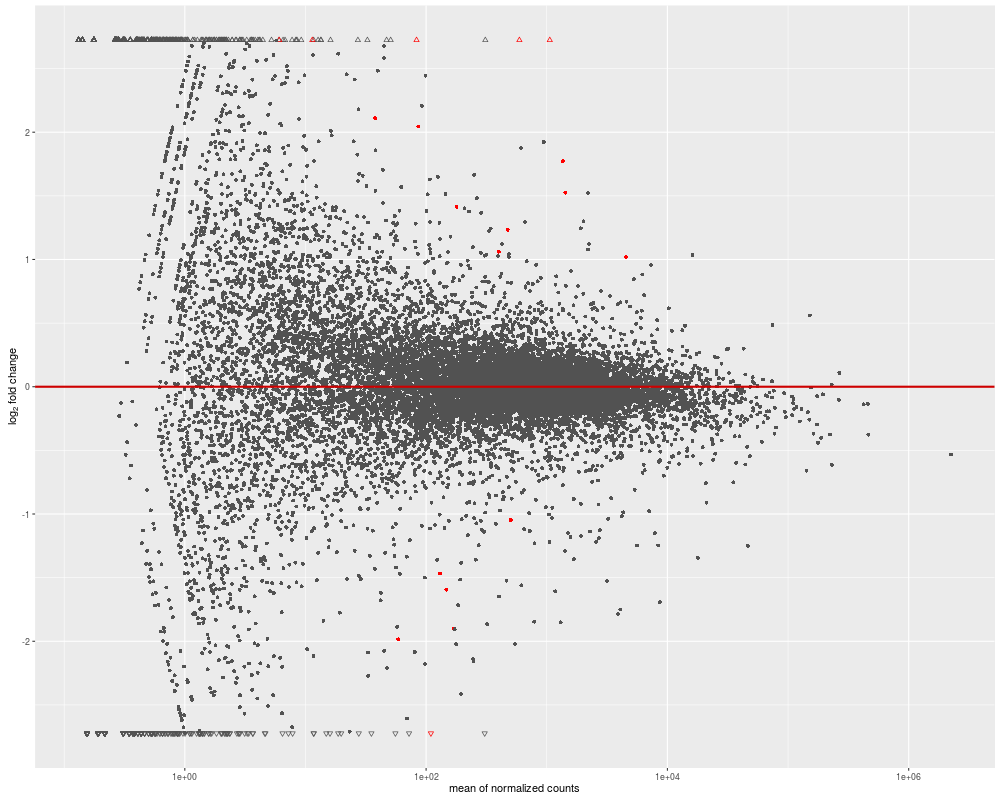
No, you absolutely can't. High/low log2 fold changes are highly biased toward lowly expressed genes, as you can see in the typical MA-plot below. However, the measure of the expression of those genes is hampered with proportionaly more noise than for highly expressed genes. Such high log2FC doesn't usually reflect a biological reality, hence the need for a treshold on the FDR/p-value that will usually filter out those lowly expressed genes with high log2FC.
There are two main differences between the Kallisto-sleuth pipeline and the 'Ballgown' pipeline.
Sleuth makes use of Kallisto's bootstrap analyses in order to decompose variance into variance associated with between sample differences and variance associated with quantificaiton uncertainty. I don't believe ballgown accounts for uncertainty in the transcript quantification.
The second difference is the quantifier. The ballgown pipeline uses Cufflinks for quantification by default, where as obviously kallisto-sleuth uses kallisto. I've not had good experiences with Cufflinks' quantification accuracy, whereas Kallisto's seems pretty good to me.
If Kallisto-sleuth gives you results, why not just use that?
If you really wanted to carry on using Ballgown, I suggest quantifying with RSEM rather than Cufflinks, which I think is possible.
Are you experienced with ballgown? I recently had a look at it and in its documentation, they state:
These models are conceptually simialar to the models used by Smyth
(2005) in the limma package. In limma, more sophisticated empirical
Bayes shrinkage methods are used, and generally a single linear model
is fit per feature instead of doing a nested model comparison, but the
flavor is similar (and in fact, limma can easily be run on any of the
data matrices in a ballgown object).
I have not gone any further in the documentation so far, but is there any advantage in using ballgown over limma or other approaches, especially because it still uses FPKM rather than more sophisticated normalization methods?
I've not really used the ballgown pipeline beyond playing about with it. My own preferred pipeline is salmon -> tximport -> DESeq2, although I can see the conceptual advantages in using sleuth. If I want an experiment specific transcript annotaiton, I'll assemble that first with stringtie and then pass it to salmon. I don't really see any statistical advantages to ballgown, although I guess the visualization and exploration tools could be useful.




Thanks everyone !! your answers are insightful and help me to pursue in a meaningful direction.