
Hello everyone,
I've been reading up and searching on forums for something that could help me interpret the fold change plots I am getting after performing differential expression analysis on my data. I think that was worse because I think I've made myself start second guessing my analysis, so I've decided to ask for some other opinions. I'll have to be a bit vague about the specific treatments and organism because of NDA agreements with the financing entity, but otherwise I think it should be fine.
Some context: we have two treatments for our organism (B and C) and are analyzing the expression in four different tissues (G, M1, M2, M3), and we did this in triplicate, for a grand total of 24 samples. Since our organism is not sequenced, we had to do de novo assembly with Trinity on pooled read data to create a reference (around ~560 million reads). We have around 300,000 assembled transcripts, but calculating ExN50 and TPM, less than 35,000 transcripts account for about 96% of the data in contigs with an N50 of almost 2 kb. The vast majority of assembled transcripts have 5 or less assigned reads.
I used Salmon to calculate abundance for all samples against the reference, and then used TxImport to pass the data for analysis with DESeq2. For DESeq2, the two factor combinatorial design was built into ~group so we could later extract contrasts between tissues within one treatment, or between equivalent tissues between treatments.
dds <- DESeqDataSetFromTximport(txi.salmon, colData=colDesign, design= ~tissue+treatment)
dds$group <- factor(paste0(dds$treatment,dds$tissue))
design(dds) <- ~group
de_results <- DESeq(dds, parallel=TRUE, BPPARAM=MulticoreParam(2))
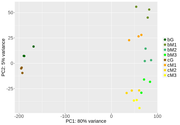
After this, I've been doing some exploratory analysis. First, PCA analysis after looks good. Samples from different tissues cluster away from one another (quite drastically in G vs the M tissues), and there is some smaller separation in the same tissue with different treatment. So far so good.
However, when I start looking at the volcano plots or MA plots, they look rather atypical. It seems that the nature of the data does not allow to detect small fold changes. The vertical lines mark -2 and 2 fold. All the changes determined as significant are of a large magnitude. Even the glut of transcripts with very few reads that show up in the MA plot tend to have large fold changes that are not significant. I don't know if this should worry me. Also, some comparisons that have this odd vertical line of significant values. I've normally gotten volcano plots with data more spread out, this feels like an artefact.
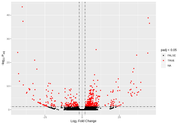
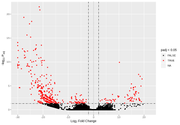
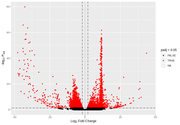
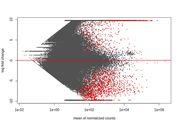
Any tips as to something that might be wrong when setting up the DESeq2 design, or ideas about the data itself are quite welcome.

For what it's worth, you appear to be doing everything correctly. I wonder if part of the oddness of some of the plots comes from the difficulties in transcriptome assembly. This is a bit of a shot in the dark, but have a look at the Lace package from Alicia Oshlack's group. It should be able to build "super transcripts", which I strongly suspect will be a much more useful thing to quantify.
Also, are G, M1, M2, and M3 really tissues or are these cell cycle phases?
It does give me some peace of mind to know I'm not faulty in my steps. The G and M are different tissues from different parts of the body. We have some information that indicates that the treatments will affect different organs. Therefore, it wasn't entirely surprising to have quite a large separation by PCA. I will check out the Lace. We do want to explore if there is some isoform change between the M samples (with all the caveats of Illumina sequencing for such things, we already have the data), so that will be fused by this tool, if I understand the general purpose correctly.
Yes, I think Lace will at least partially fuse isoforms, though it'd make sense to then abandon it once you look at isoform switching.
I did go through with the Trinotate workflow to assign some functional annotation to the transcripts. After some filtering, I can get around 45000 transcripts for which I can retrieve some homology, ORF prediction and/or PFAM domain. I don't think chimeric contigs are the issue, the length distribution doesn't look weird. Most contigs are these small 200-300 assemblies, and only a few are assembled into contigs larger than 3 kb.
In any case, at this point I think it might be more of a case of including far too many uninformative transcripts into the model. I used the plotDispEsts() function of DESeq2 on the model object, and sure enough it's not looking great.
Do you suggest asking over at Bioconductor support for some other input?
Did you try it without G-phase samples? The difference between G- and M-phase samples is 'extraordinary'. 80% on a PC1 in PCA analysis is a huge difference, and I would fully expect that to distort the statistical analyses.
I have been toying around with parameters and settings in the model. Following your suggestion I did use either only the G or the M samples. The PCA plots of both show that the B treatment has higher dispersion than the C treatment, which cluster nicely together. DESeq2's one parameter for within group variation might be inadequate.
Contrasts with the same tissue between B and C treatment resulted in a hill shaped p-value distribution. It doesn't really change if I do it with all the samples together, or with just the G or M samples. Following the recommendations of Klaus and Huber I took the steps to re-estimate the null model dispersions with all the samples using fdrtool. I did manage to flatten out the p-value distributions. Well, most of them. Some still show a peak at 1 that almost disappears if I filter by higher base mean values.
Before FDR
After FDR
After the FDR correction, Volcano plots look more "conventional", but in some contrasts the number of significant transcripts rises a lot, from a few hundred or around a thousand to tens of thousands.
Thinking that it might be small count transcripts were being called as significant, I ran DESeq with the betaPrior= TRUE argument to perform LFC shrinkage. I did try to use the lfcShrink() function after running the results(), but it ran for hours on a single contrast and then failed, so I'm not sure what the problem is there. From what I understand there is a slight difference in this procedure (besides future-proofing for different shrinkage estimators), in how the p-values are computed, towards the shrunken LFC or the un-shrunken values. Although doing the shrinking and the fdrtool procedure are correcting for different things, I am not exactly sure if they are statistically compatible (or compatible with including a lfc threshold in the results() function with an alternate hypothesis ).
This organism does not have any database information of any kind. The closest thing is another species of the same family whose transcriptome was also recently obtained and analyzed in broadly the same way. Neither has a sequenced genome or curated gene model.
That does make it more difficult.
What if you predict which transcripts have the capability to encode protein-coding genes?
CPAT is one program to predict coding potential, but there are others.
Also, what is the length distribution for your transcript assembly? Last time I tested a Trinity assembly, I thought it tended to give transcripts that were incorrectly joined (which would cause there to be more long transcripts). If I wanted to test for the species of origin, I would use Oases. I think those transcripts / contigs tend to be smaller. However, this may further reduce the total number of coding genes (since I would say you are more likely to have partial sequences), but it would provide another option to test estimations and fold-change calculations.
You could also see the effect of changing parameters for the Trinity and/or Oases assembly.
Finally, are there any transcripts in NCBI Nucleotide (or even a partial set of sequences in RefSeq)? Filtering and annotation may take a little while, but even a partial set (perhaps a few thousand genes for a Eukaryote?) might be something worth considering (and, if your coverage / fold-change improves, having information about gene function could be helpful).