
Hi,
I am working on an RNA-Seq project. We just got back some sequencing and I had a couple questions about it. I figure someone here might be able to help me out a bit.
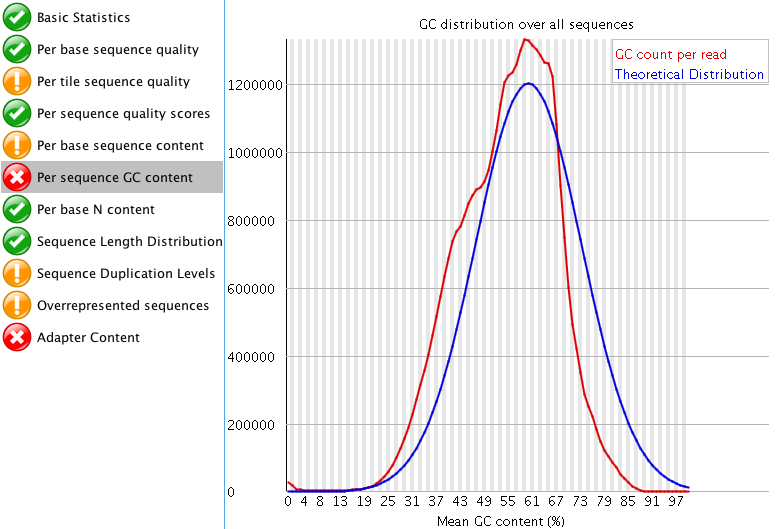
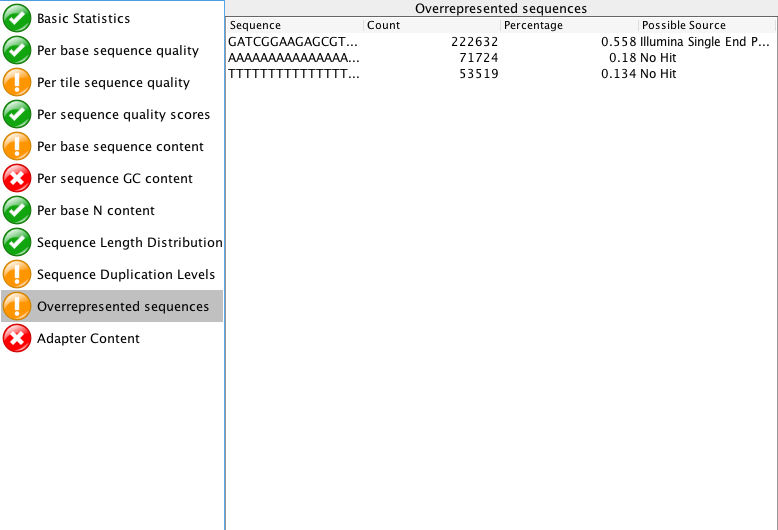
So I think the sequencing doesn't look too bad. I don't really like trimming RNA-seq data if I don't have to, but I attached images of my FASTQC results.
The "Per tile sequence quality" makes me think that I may need to trim that data a bit? What do you think? I am also curious what people think about the "Adaptor content"? The read length for this project is 150 bp. Do you think those are just reads started to end at ~60 bp instead of the full 150 bp?
Let me know if you see anything else that really stands out for you. Again, I would prefer to do as little amount of trimming as possible.. if I even have to.
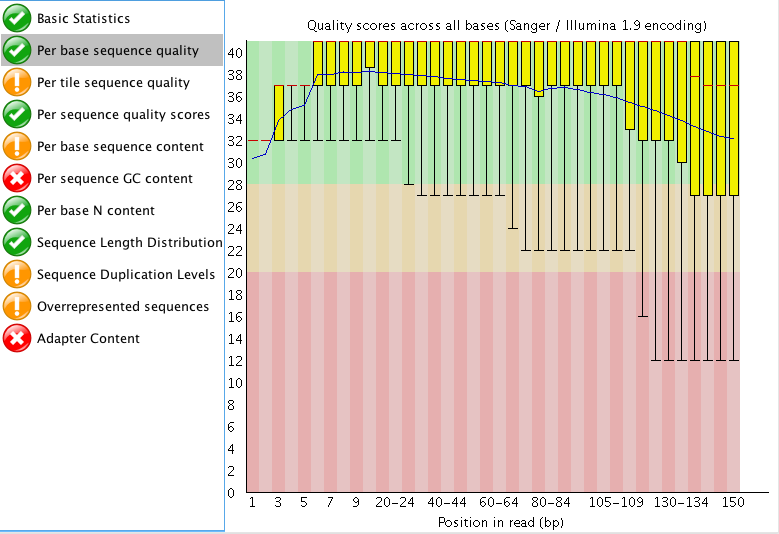
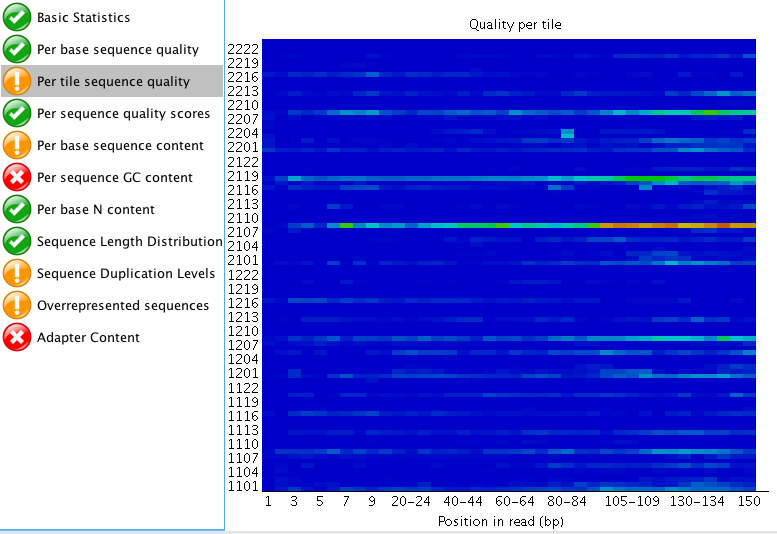
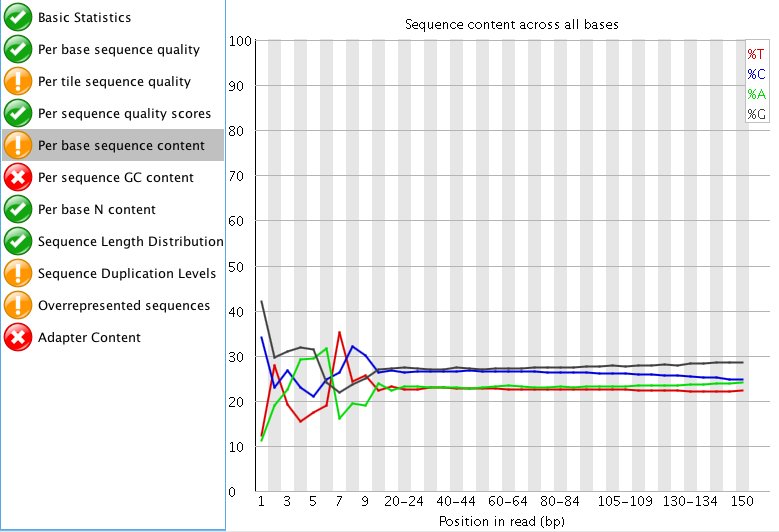
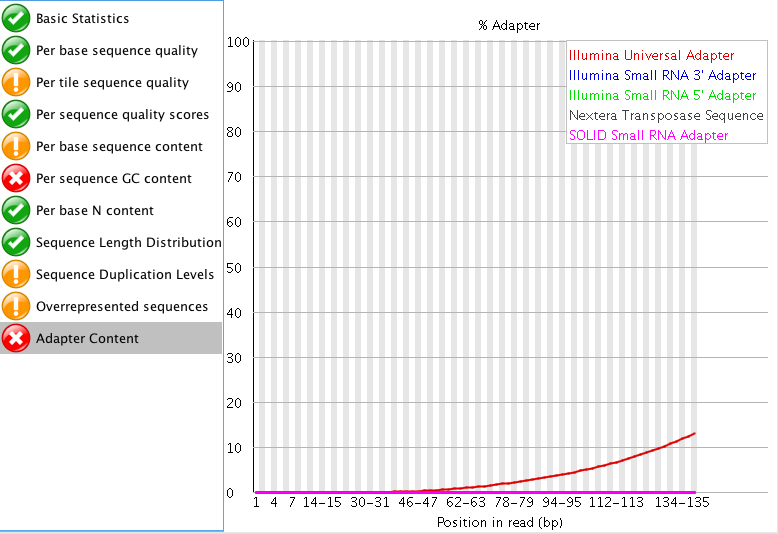
"Per tile sequence quality" is just the physical layout on the flow cell. There is not much you can do about this and it doesn't look worrying to me. I'd just trim adapters and polyA/T tails and stop there.
Agreed. Much more important for me is that I get solid downstream results like adequate % of mapped reads, reasonable correlations between replicates and PCA clustering of samples that should cluster together based on its biological background.
When you say adapters, are you referring to the "Overrepresented sequences"? or the actual "Adapter content"?
Also, why do you think there are single end adaptors? This was a paired end?