
Hello everyone,
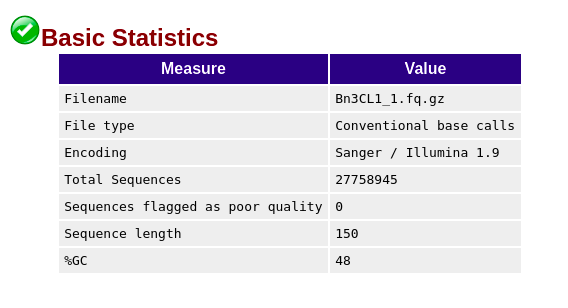
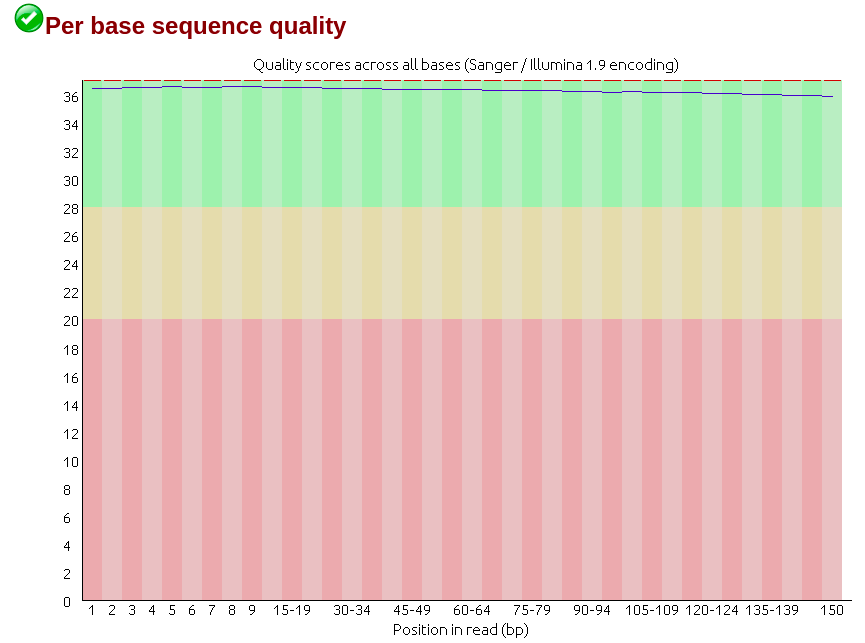
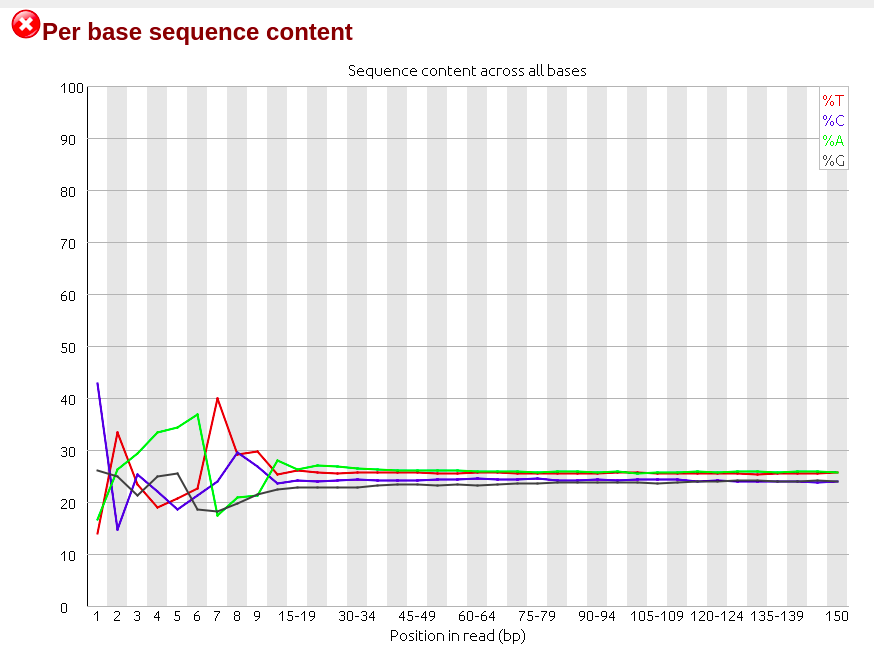
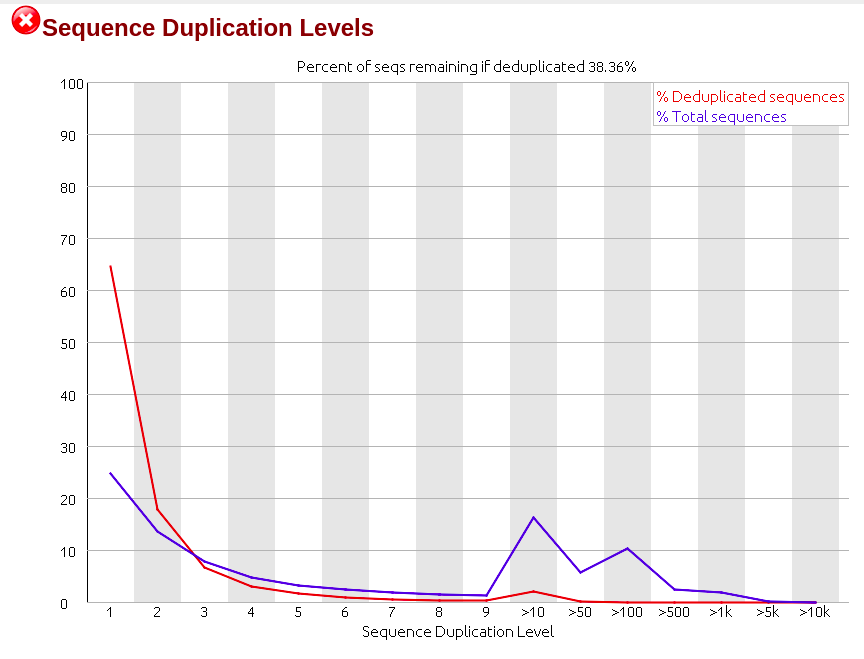
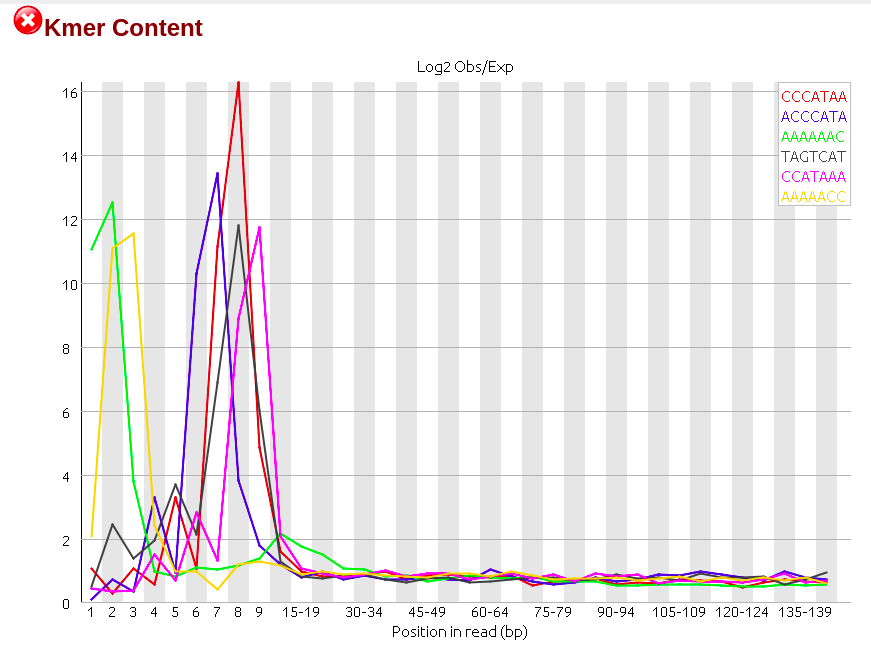
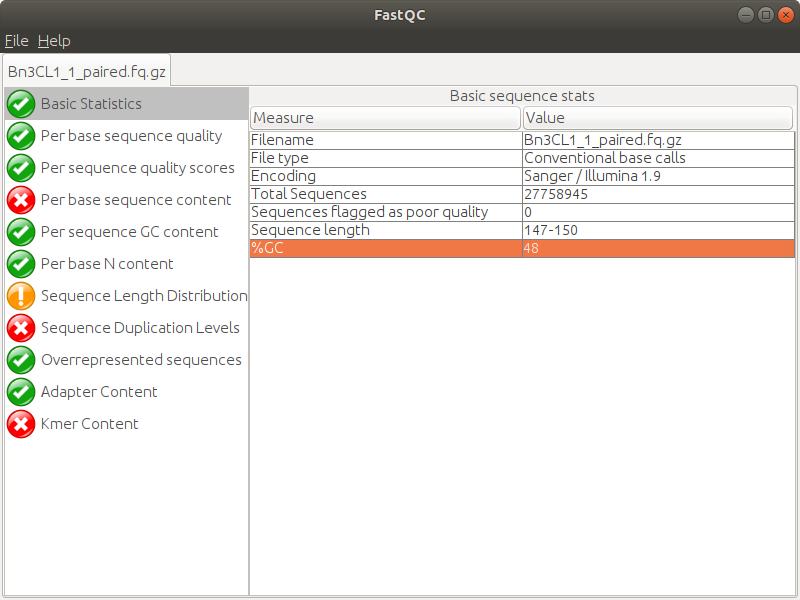
Days ago I just got RNA-seq data which is Illumina paired-end library where reads are 150bp in length, and did quality control via Fastqc (version 0.11.5), finding though the sequencing quality is really good there are several failures within the results mainly on three aspects: 'Per base sequence content', 'Sequence duplication level' and 'Kmer content'. (The detail of my failure I'll present below as pics)
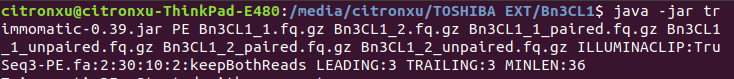
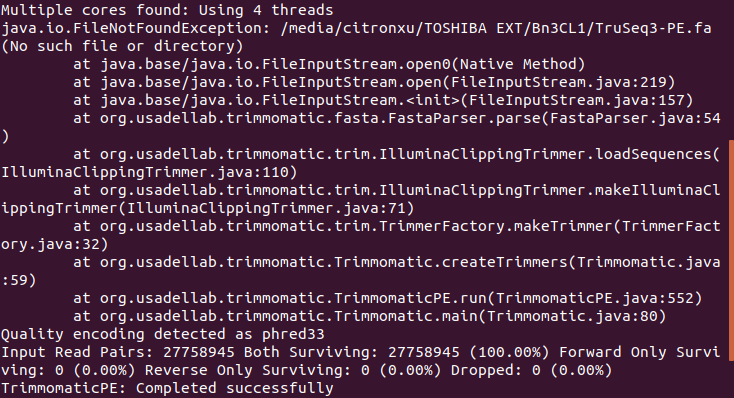
Then I realized that among all failures, the consensus is always the first nine to ten nt causing the quality dropping down dramatically with results on following nt position being quite good. So I raised a hypothesis, if the raw data received from company would be yet going through adapter-trimming process? Afterwards, I run trimmomatic to see what would be gonna happen to the reads. Surprisingly when I run Fastqc on its outcome there is big change on the final results where parts of my reads were clipped by 3 nt ending up with 147bp in length.
Here is my question, how can it happen some failures occurring in my datasets even under such a good quality? And does the quality report of trimming tell actually the original data from the company has already been trimmed and could be used for downstream analysis?

Thanks a lot!!! perfectly solved my question.
do note the error message, you are not actually trimming the adapters,
trimmomatic will chug along even when the file is missing then cheerfully reports "completed successfully" when in fact there was a major error of a missing adapter file
in general I would recommend running the
SLIDINGWINDOWoperation,it really makes no sense to to do
TRAILING 3a quality of 3 is just as bad as quality 4, 5 or 6 - you should do a sliding window say 30 average over 4-5 bases or something similar..