
Note: The original title is "Why Salmon produces very different quantification results compared with featureCounts for lncRNA genes?" But later I found that if I run salmon with all transcripts but not only with protein-coding or lncRNA genes, the correlation between featureCounts and salmon became higher.
Hello, recently I analyzed about 20 RNA-seq samples. I adopted two approaches to quantify the expression level of genes.
STARmapping ->featureCounts(only use uniquely mapping reads)salmonquantification -> summarize isoform-level expression into gene-level bytximport
I compared the quantification results from two methods, and calculated the correlation of coding genes and lncRNA genes, separately. The table showed the correlation of each sample (only listed 5 samples, NOTE: coding transcripts fasta and lncRNA fasta were used by salmon for quantifying, seperately):
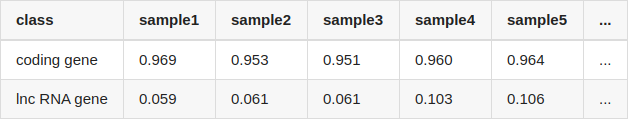
The quantification results of salmon and featureCounts correlate very well for coding genes, but for lncRNA genes, the correlation of them is extremely low.
Table Update: I've mentioned that I quantified lncRNA and protein coding gene using salmon. But I may have used inappropriate transcript fasta files for quantification: the lncRNA gene and protein coding gene were quantified with gencode.v34lift37.pc_transcripts.fa and gencode.v34lift37.lncRNA_transcripts.fa, separately. If I use all transcripts (gencode.v34lift37.transcripts.fa), the results became quite different:
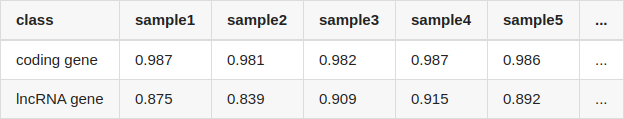
Though the correlation (Pearson correlation) of lncRNA is still lower than that of protein coding gene, it is no longer so large.

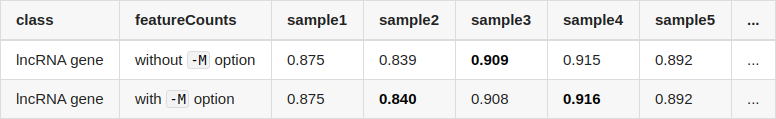

Sry for the simple question, but did you maybe use different gtf file versions for the runs?
Thank you for reminding me of this, I think the reason is that I quantified transcripts with protein-coding transcripts and lncRNA transcripts separately. If I run
salmonwith all transcripts, the difference between coding/lncRNA gene become smaller.