Entering edit mode

4.4 years ago
Sujata
▴
20
Single run is used for counting or differential gene expression, while paired-run is used for splice variants, mutations etc. Why?

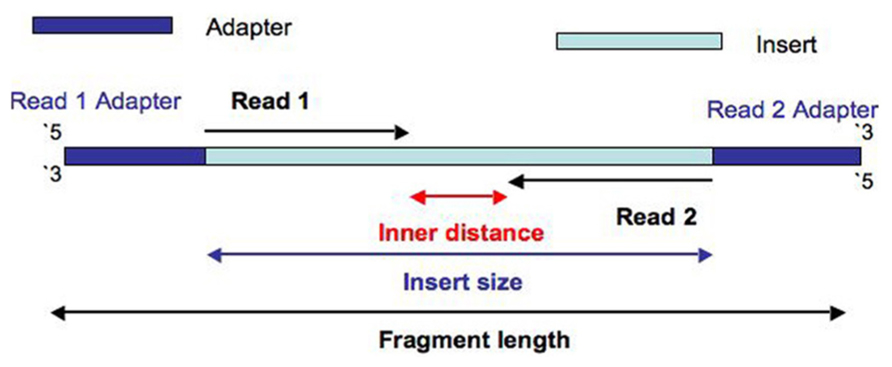
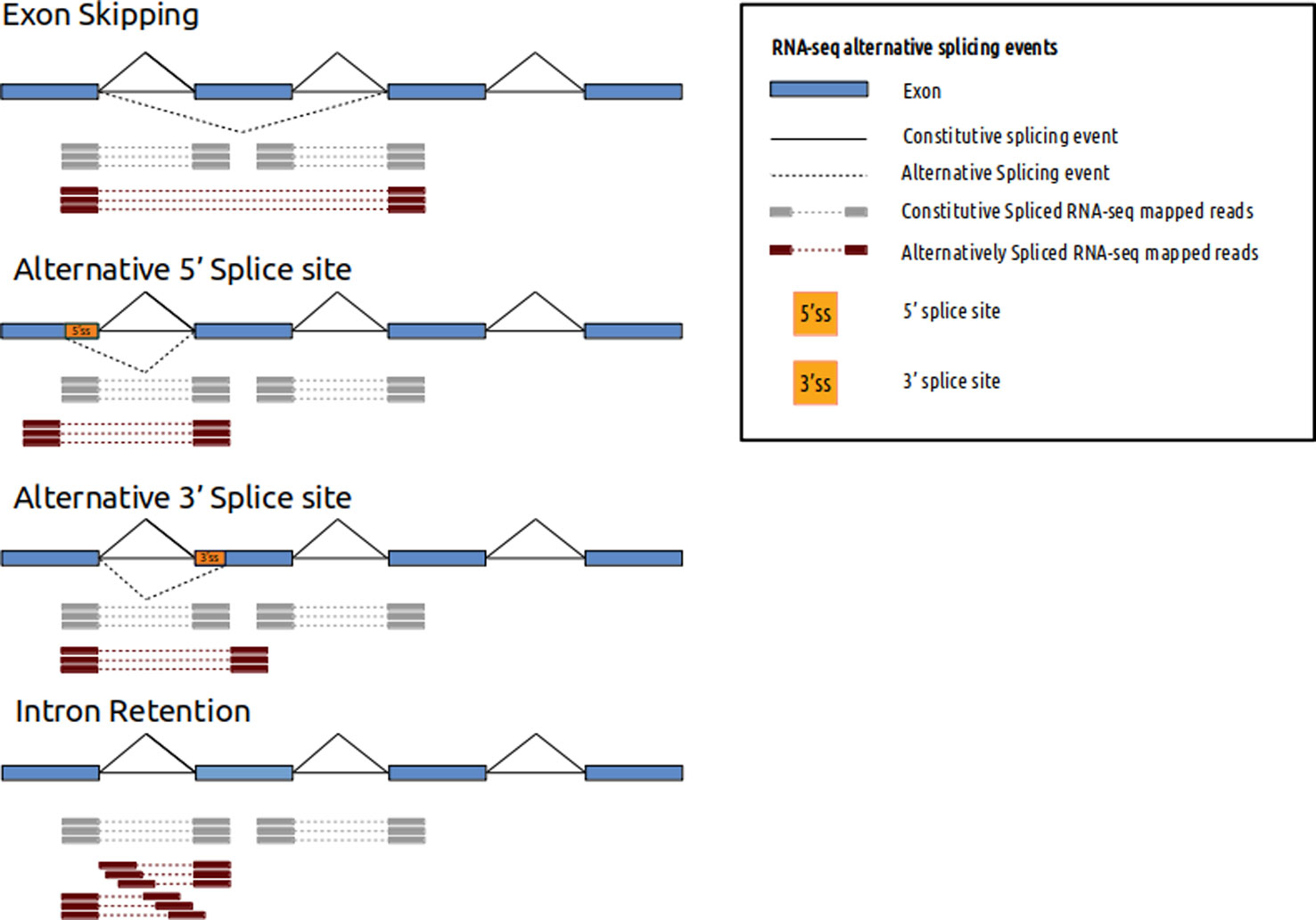
Single versus Paired-end
So, if a single-end reads spans across two exons, will it not be possible to figure out the same here?
It should be possible to do that. Only difference is here you are going to be looking at say 50-100 bp read. Where as with a paired-end data you are sampling a 350-450 bp fragment.