|
#!/usr/bin/env python |
|
|
|
from collections import namedtuple, defaultdict |
|
from pybedtools import BedTool |
|
import argparse |
|
|
|
Point = namedtuple('Point', ['id', 'pos', 'type']) |
|
Interval = namedtuple('Interval', ['chrom', 'start', 'end']) |
|
|
|
|
|
def report_interval(chrom, start, end, num_files, files_with_interval): |
|
print "\t".join([chrom, str(start), str(end), str(len(files_with_interval.keys()))]), |
|
for i in range(0,num_files): |
|
if i in files_with_interval: |
|
print "\t1", |
|
else: |
|
print "\t0", |
|
print |
|
|
|
|
|
def merge(file): |
|
""" |
|
Merge features in a BED/GFF/VCF into non-overlapping intervals |
|
""" |
|
start = -1 |
|
end = -1 |
|
chrom = None |
|
for feature in BedTool(file): |
|
if feature.start - end > 0 or end < 0 or feature.chrom != chrom: |
|
if start >= 0: |
|
yield Interval(chrom, start, end) |
|
start = feature.start |
|
end = feature.end |
|
chrom = feature.chrom |
|
elif feature.end > end: |
|
end = feature.end |
|
yield Interval(chrom, start, end) |
|
|
|
|
|
def load_and_sort_points(files): |
|
""" |
|
""" |
|
file_id = 0 |
|
chrom_points = defaultdict(list) |
|
# for each input file, first merge the features into non-overlapping |
|
# intervals using merge(). Each non-overlapping feature is then |
|
# broken up into discrete "Points": one for the start and one for the end. |
|
for file in files: |
|
# merge the file and split features into points |
|
for feature in merge(file): |
|
s = Point(file_id, feature.start, "start") |
|
e = Point(file_id, feature.end, "end") |
|
chrom_points[feature.chrom].append(s) |
|
chrom_points[feature.chrom].append(e) |
|
file_id += 1 |
|
|
|
# sort the points in for each chrom |
|
for chrom in chrom_points: |
|
chrom_points[chrom].sort(key=lambda i: i.pos) |
|
return chrom_points |
|
|
|
|
|
def load_genome(genome): |
|
chrom_sizes = {} |
|
for line in open(genome, 'r'): |
|
fields = line.strip().split("\t") |
|
if len(fields) > 1: |
|
chrom_sizes[fields[0]] = fields[1] |
|
|
|
return chrom_sizes |
|
|
|
|
|
def nway(files, genome): |
|
""" |
|
Assumptions: input files must contain non-overlapping intervals |
|
|
|
1. Example using already-merged files: |
|
$ cat a.merged |
|
chr1 6 20 |
|
chr1 22 30 |
|
|
|
$ cat b.merged |
|
chr1 12 32 |
|
|
|
$ cat c.merged |
|
chr1 8 15 |
|
chr1 32 34 |
|
|
|
|
|
$ ./nway-cluster.py a.merged b.merged c.merged |
|
#chr st ed num a b c |
|
chr1 0 6 0 0 0 0 |
|
chr1 6 8 1 1 0 0 |
|
chr1 8 12 2 1 0 1 |
|
chr1 12 15 3 1 1 1 |
|
chr1 15 20 2 1 1 0 |
|
chr1 20 22 1 0 1 0 |
|
chr1 22 30 2 1 1 0 |
|
chr1 30 32 1 0 1 0 |
|
chr1 32 34 1 0 0 1 |
|
|
|
|
|
2. Example using un-merged, yet sorted files: |
|
$ cat a.bed |
|
chr1 6 12 |
|
chr1 10 20 |
|
chr1 22 27 |
|
chr1 24 30 |
|
|
|
$ cat b.bed |
|
chr1 12 32 |
|
chr1 14 30 |
|
|
|
$ cat c.bed |
|
chr1 8 15 |
|
chr1 10 14 |
|
chr1 32 34 |
|
|
|
$ ./nway-cluster.py a.bed b.bed c.bed |
|
#chr st ed num a b c |
|
chr1 0 6 0 0 0 0 |
|
chr1 6 8 1 1 0 0 |
|
chr1 8 12 2 1 0 1 |
|
chr1 12 15 3 1 1 1 |
|
chr1 15 20 2 1 1 0 |
|
chr1 20 22 1 0 1 0 |
|
chr1 22 30 2 1 1 0 |
|
chr1 30 32 1 0 1 0 |
|
chr1 32 34 1 0 0 1 |
|
|
|
|
|
3. Thanks to pybedtools, it works with BAM files as well. |
|
But I hope you have a machine with lots of RAM. |
|
./nway-cluster.py 1.bam 2.bam 3.bam |
|
|
|
""" |
|
num_files = len(files) |
|
|
|
# 1. load each point from each interval in each file into |
|
# a hash keyed by chrom. |
|
# 2. sort the points in asecnding order for each chrom |
|
chrom_points = load_and_sort_points(files) |
|
if genome is not None: |
|
chrom_sizes = load_genome(genome) |
|
|
|
# 3. Rip through the points and find shared intervals |
|
for chrom in chrom_points: |
|
files_with_interval = {} |
|
prev_point = 0 |
|
for point in chrom_points[chrom]: |
|
# report the current interval if we've moved at all along the chrom. |
|
if point.pos > prev_point: |
|
report_interval(chrom, prev_point, point.pos, num_files, files_with_interval) |
|
# if we're at a start, we add the current file to the active list of files. |
|
# otherwise, an end point means we can drop the current file. |
|
if point.type == "start": |
|
files_with_interval[point.id] = 1 |
|
else: |
|
del files_with_interval[point.id] |
|
prev_point = point.pos |
|
|
|
# if requested, handle the interval from the last observed point to the end of the chrom |
|
if genome is not None and point.pos < chrom_sizes[chrom]: |
|
report_interval(chrom, point.pos, chrom_sizes[chrom], num_files, files_with_interval) |
|
|
|
|
|
def main(): |
|
parser = argparse.ArgumentParser(prog='nway-cluster') |
|
parser.add_argument('files', metavar='FILE', nargs='+', |
|
help='***merged*** (non-overlapping intervals) BED files to intersect') |
|
parser.add_argument('-g', metavar='GENOME', dest='genome', default=None, |
|
help='The \"genome\" file: i.e., a list of chroms and their sizes.') |
|
|
|
args = parser.parse_args() |
|
nway(args.files, args.genome) |
|
|
|
if __name__ == "__main__": |
|
main() |


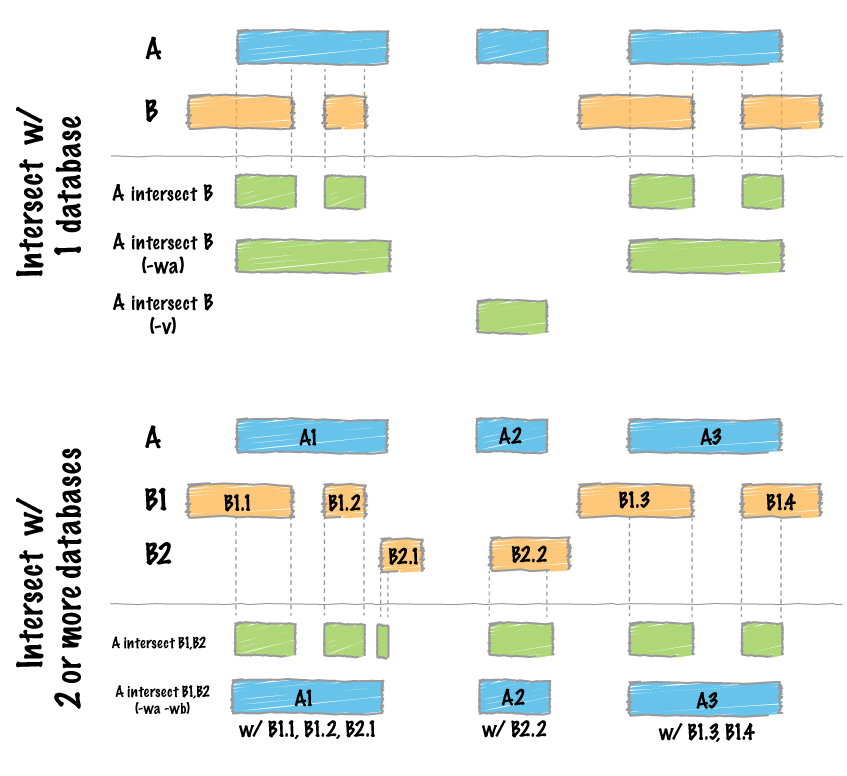

+1. Great example of how questions on biostar are helping stimulate advances in bioinformatics technology.
agreed. it was quite fun to write.
Hi Aaron, Is the
-fparameter still functional in MultiIntersectBed, if I need a minimum overlap of 50% in all the three files.Thanks
oh cannot thanks more..
Might be a very naive question, Well I have 5 peak files and the sum of peaks is 100000 but when I use multiIntersectBed I get a total number of peaks to be 150000 why such a big difference? Does the script breaks the total regions into some bins and then makes an intersection?, if yes. what is the size of these bins?
Almost exactly what I'm looking for. Is there a way to also print out all the features from each of the 3 bed files?
Thanks.
-emptyIs multiinter strand-aware? The output doesn't give any clues whether it is!