Entering edit mode

7.4 years ago
sacha
★
2.4k
Hi,
I recoded fastQC application from Java to Qt/C++ just for fun :D .
It should be faster, but I didn't make a real benchmark.
It was really easy to port the java code to Qt . In most of the time I just did a copy paste! Of course, I did some changes to keep C++/Qt pattern rule.
Currently it can load only Fastq file... I will make Fastq.gz working as soon as possible.
And there are only 4 kind of analysis . I only make my favorite analysis right now. If you need one, feel free to ask me.
Code source available here : https://github.com/labsquare/fastQt
License GPL like FastQC.


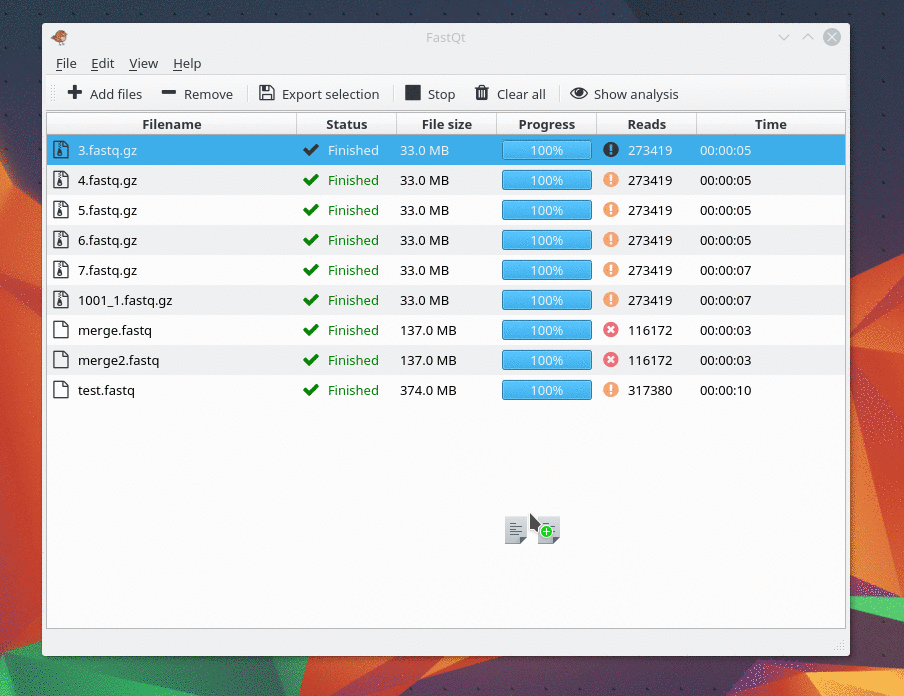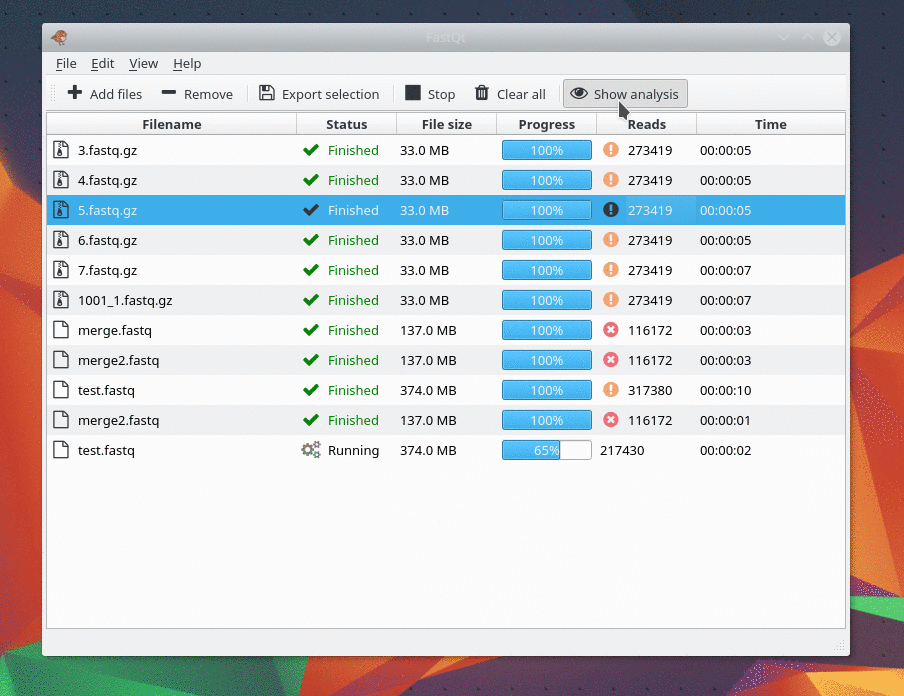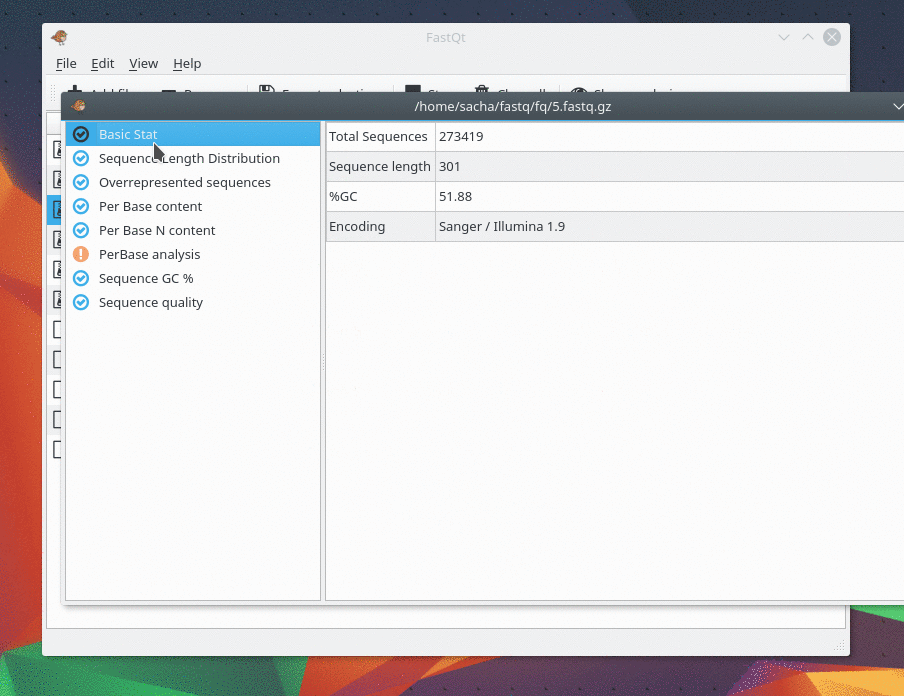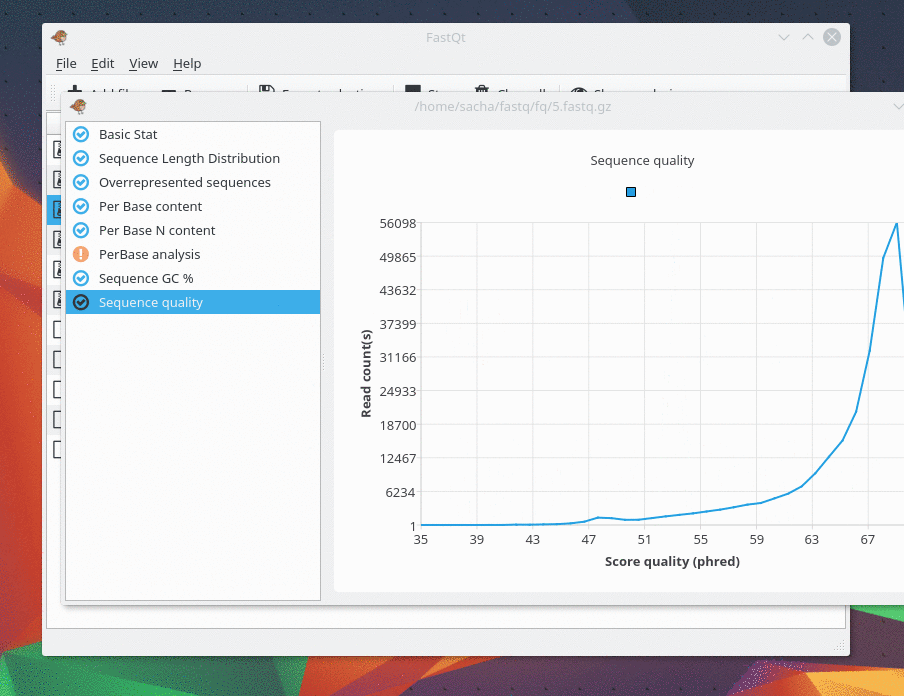


Version 0.1 released !
I m pleased to present you the first release of FastQt version 0.1 . This version is pretty similar than FastQC and probably faster . It supports fastq, fastq.gz, fastq.bz2 and fastq.xz . Be free to tell us if you have bugs or any suggestion. The goal of the 0.1 version was to have same features than fastQC. The next goal in version 0.2 will focus on improvement and new feature like "multi analysis" in the same view and "Console mode". You can Download source code and compile it under Linux by following this link : https://github.com/labsquare/fastQt
A Binary for Linux x86_64 can be found here .
You just have do download it and run it as follow:
Labsquare Team !
Is there a size limit to the reads? I assume the tool isn't suited for "third-generation sequencing" data such as PacBio and Oxford Nanopore?
sequences size you mean ? I tested yesterday a fastq with long reads. It was working. Give me some data, I will try
Yes, reads of multiple kb. And note that those reads will have very different sizes in the same dataset. Some human WGS nanopore sequencing data is available from https://github.com/nanopore-wgs-consortium/NA12878
Hi,
I am a contributor of fastqt and I work on long-reads every day, it's work very wheel and I have a feeling it's faster than fastqc.
Thank for your interest.
Great, thanks for clarifying!