I answered my own question by finding this http://tolstoy.newcastle.edu.au/R/e9/help/10/02/6212.html post in the R-help mailing list. If you replace the corresponding blocks in heatmap.2 with blocks of code provided in that post you can get multiple color side bars in heatmap.2. See http://tolstoy.newcastle.edu.au/R/e9/help/10/02/6307.htm for more customizations.
UPDATE2: https://github.com/nachocab made further improvements that allows a 1-dimensional matrix for the annotations, and fixed the NA problem in the color key. @amcdavid contributed a further fix to the former.
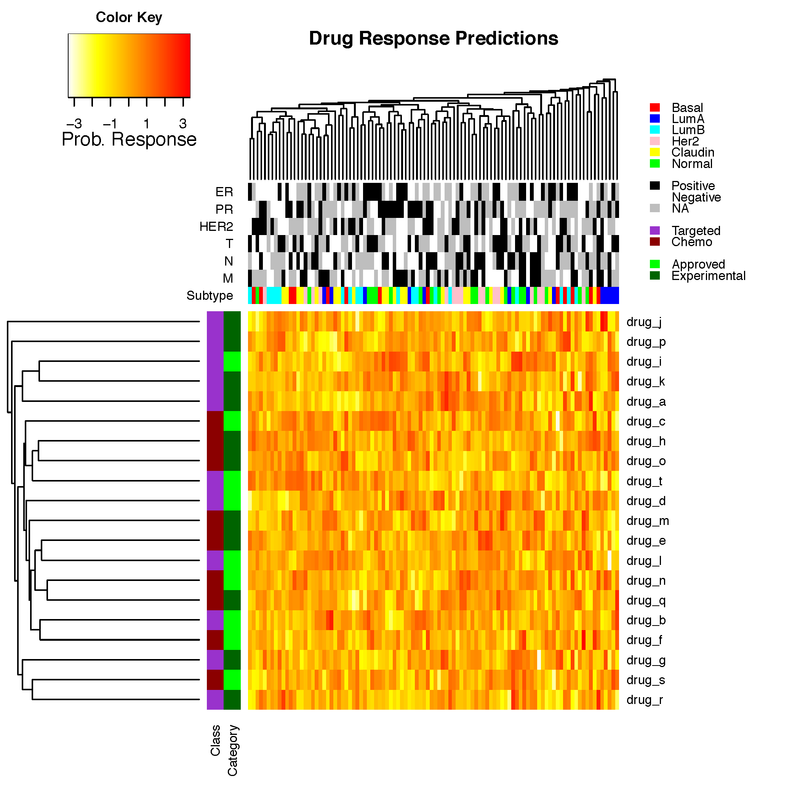
UPDATE3: I have tidied this post up a bit. Added back image which got lost along the way. Here is the latest code (github). You can just source this file and then call the heatmap.3 function with the new options as in the below example.
#If not already installed
#install.packages("gplots")
#install.packages("devtools")
#Load necessary packages
library("gplots")
library("devtools")
#Load latest version of heatmap.3 function
source_url("https://raw.githubusercontent.com/obigriffith/biostar-tutorials/master/Heatmaps/heatmap.3.R")
#Set a working directory for output files
setwd("/Users/ogriffit/git/biostar-tutorials/Heatmaps")
#Create a fake dataset for demonstration purposes
prob_matrix=replicate(100, rnorm(20))
drug_names=paste("drug",letters[1:20],sep="_")
patient_ids=paste("patient",c(1:100),sep="_")
rownames(prob_matrix)=drug_names
colnames(prob_matrix)=patient_ids
#Create fake color side bars
drugclass_colors=sample(c("darkorchid","darkred"), length(drug_names), replace = TRUE, prob = NULL)
drugcategory_colors=sample(c("green","darkgreen"), length(drug_names), replace = TRUE, prob = NULL)
subtype_colors=sample(c("red","blue","cyan","pink","yellow","green"), length(patient_ids), replace = TRUE, prob = NULL)
Mcolors=sample(c("black","white","grey"), length(patient_ids), replace = TRUE, prob = NULL)
Ncolors=sample(c("black","white","grey"), length(patient_ids), replace = TRUE, prob = NULL)
Tcolors=sample(c("black","white","grey"), length(patient_ids), replace = TRUE, prob = NULL)
HER2colors=sample(c("black","white","grey"), length(patient_ids), replace = TRUE, prob = NULL)
PRcolors=sample(c("black","white","grey"), length(patient_ids), replace = TRUE, prob = NULL)
ERcolors=sample(c("black","white","grey"), length(patient_ids), replace = TRUE, prob = NULL)
rlab=t(cbind(drugclass_colors,drugcategory_colors))
clab=cbind(subtype_colors,Mcolors,Ncolors,Tcolors,HER2colors,PRcolors,ERcolors)
rownames(rlab)=c("Class","Category")
colnames(clab)=c("Subtype","M","N","T","HER2","PR","ER")
#Define custom dist and hclust functions for use with heatmaps
mydist=function(c) {dist(c,method="euclidian")}
myclust=function(c) {hclust(c,method="average")}
#Create heatmap using custom heatmap.3 source code loaded above
pdf(file="heatmap3_example.pdf")
main_title="Drug Response Predictions"
par(cex.main=1)
heatmap.3(prob_matrix, hclustfun=myclust, distfun=mydist, na.rm = TRUE, scale="none", dendrogram="both", margins=c(6,12),
Rowv=TRUE, Colv=TRUE, ColSideColors=clab, RowSideColors=rlab, symbreaks=FALSE, key=TRUE, symkey=FALSE,
density.info="none", trace="none", main=main_title, labCol=FALSE, labRow=drug_names, cexRow=1, col=rev(heat.colors(75)),
ColSideColorsSize=7, RowSideColorsSize=2, KeyValueName="Prob. Response")
legend("topright",legend=c("Basal","LumA","LumB","Her2","Claudin","Normal","","Positive","Negative","NA","","Targeted","Chemo","","Approved","Experimental"),
fill=c("red","blue","cyan","pink","yellow","green","white","black","white","grey","white","darkorchid","darkred","white","green","darkgreen"), border=FALSE, bty="n", y.intersp = 0.7, cex=0.7)
dev.off()
#Example to show that it now also works with just a single column or single row
mat <- matrix(1:100, byrow=T, nrow=10)
column_annotation <- sample(c("red", "blue", "green"), 10, replace=T)
column_annotation <- as.matrix(column_annotation)
colnames(column_annotation) <- c("Variable X")
row_annotation <- sample(c("red", "blue", "green"), 10, replace=T)
row_annotation <- as.matrix(t(row_annotation))
rownames(row_annotation) <- c("Variable Y")
heatmap.3(mat, RowSideColors=row_annotation, ColSideColors=column_annotation)





Thanks! That's really useful. BTW, could you provide a complete example to exactly reproduce your figure?
This should have been a comment. Not an answer. I have provided example R code. Unfortunately I am not able to provide a complete example because the data underlying this particular figure is very large and not available for public posting yet.
I found some time and re-worked my answer above to provide a complete working example, now on random data, rather than data I couldn't release here.
cited in : "SEER and Gene Expression Data Analysis Deciphers Racial Disparity Patterns in Prostate Cancer Mortality and the Public Health Implication" https://www.nature.com/articles/s41598-020-63764-4
Hi,
I have a question about the script above. When I run it I get the error message:
I read a lot but I cannot find any solution. Has anybody an idea?
Thanks in advance.
Janita
This should have been a comment to the answer given above. You seem to have an encoding problem, does it really say
'Â'?yes it does. Do you have an idea how to fix this?
I have the same issue. Great post.
I can't guess what this might be. Other than making sure that I had gplots and devtools installed, I just copied and pasted the script below into R verbatim and it ran without error. At what point exactly does this error occur? The only thing I can guess is that it is some browser issue?
I run into the same problem too. I'm using Rstudio 0.98 and R 3.1.2.
I ran only the following code.
and got the error message
I searched a lot but can't find anything of
'Â'means. Has anyone solve this?Thanks,
Jack
I still can't really guess what is going on here. I just copied the code below into Rstudio 0.98 (R 3.1.2) and it ran without problems. If you type 'heatmap.3' I presume you see the function code has been successfully loaded into your session using source_url? Similarly, typing 'prob_matrix' produces sensible output? I'm guessing you have gplots and devtools installed and loaded or you would have gotten a different error. The only thing I can imagine is that your browser is doing something weird when you copy/paste the code from biostar and some hidden characters are coming along for the ride. Maybe try going directly to the source at github for both the function code and example script. See https://github.com/obigriffith/biostar-tutorials. You want Heatmaps/heatmap.3.R and Heatmaps/heatmap3_example.R. Safest might be to just clone the whole repo, navigate to the correct folder and then copy the function and script into R/Rstudio from a text editor you trust.
By copying the whole heatmap.3.R code fixed this problem. I think its because there is something wrong with the following code. But I'm not sure.
Thanks a lot for your help!